ADSR Perturbation Response Envelopes
I found myself staring at this the other day, bothered by something I couldn't quite articulate.
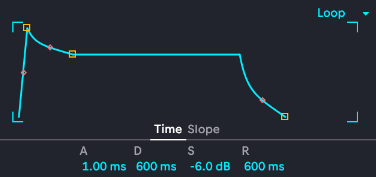
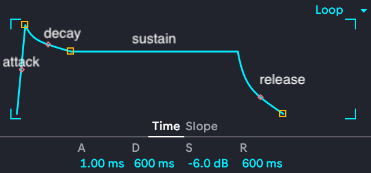
This is the ADSR envelope that's reused all over Ableton, which is a complicated digital audio workstation that lets me produce absolutely awful music at an astonishing speed using sounds no one has ever heard before. Making good music is a lot slower. Similar ADSR envelopes are used all over musical composition software, where they're used to shape the fancy note and effect parameter modulation that's all over modern music (especially electronica and its fractal subgenera). A stands for attack, it controls how fast the note actually plays to full volume once I hit it. A fast attack is instant, a slow attack will gradually swell (think Sigur Ros or Massive Attack). D stands for decay, which controls how fast the note drops from peak attack down to sustain. S for sustain is for how the note plays while you hold a note--you can set it lower than the attack to get a note that comes on strong then backs off, modeled after wind instrument players being able to use different volumes of air or bowed string players being able to press harder. Finally, R is release, which is what happens after you let go of the note: does it ring out and linger or does it abruptly stop? Here's a labeled version:
Stacking envelopes...
And you can even arrange a bunch of them together with skewed shapes to get weirder cooler sounds.
Technically the above are synthesizer input patterns instead of strictly ADSR envelopes, but I promise this will become relevant again soon.
It didn't click what was bothering me about this until much later on a walk, but once it did I couldn't un-see it: the way we talk about modulating MIDI notes in bleep bloop music is more sophisticated than the way we talk about perturbation pharmacodynamics even though the latter is a matter of life or death while the former are abused to sell cheeseburgers!
Without naming names, just think about what kind of information you get if you're evaluating a preclinical drug package that a few million dollars have been invested in: whoever's pitching will dutifully trot out their lead compound's IC50 or its equivalent1, you might get a basic plasma and urine profile on absorption, Cmax, and clearance half life, and if the team is good you'll also have a few views into the candidate's target engagement profile. Maybe some SPR kinetic plots, perhaps some Westerns showing target or downstream decreases, ideally some kind of live cell fluorescent reporter construct with validation against a scrambled control construct. If they're advanced you'll get some tissue IHC too, although we might also get a more comprehensive pharmacodynamic time course if the target is present in PBMCs. But that's about it. We can compare these attributes across animal models, we can run a series of drug-drug interaction studies in vitro with index perpetrators, we can nod seriously at the liver microsomal metabolism assay, and then we have to make hard decisions about which drug candidates to allocate scarce IND-enabling resources to. Eventually, the ones that make it through will get to their Phase 1 clinical trials, where with longitudinal post-treatment blood draws (not standard) we finally get pharmacodynamic engagement in real human tissues data and can compare it against the 2+ animal models used preclinically. When you zoom out, this is an awful thin set of parameters for making such important decisions! No wonder clinical trial attrition is so damn high!
What if, instead, we modeled each drug like this?
And then realized that we can use the same framework to also model each drug's biological effects both at the target of interest and in its interacting features (other 'omic layers, comorbidities, co-treatments, etc)? That's right, perturbation envelopes can be stacked:
Into perturbation surfaces.2
What follows is not yet formalized and is not ready for prime time, this is more a sketch for future work. However, I think that even the sketch is useful; even if we're currently missing datasets and quantitative tools to work with and compose perturbation envelopes right this second, the framework still provides interesting conceptual handles for thinking through perturbation shapes3 and their responses.
Attack ≈ onset kinetics: how fast does the perturbation reach peak engagement after dosing. Canonically this would be time to Cmax regardless of route of administration, but that just measures drug in bloodstream (or other goo). This is still fine and useful for measuring a drug's characteristics in a biological transport system. However, if what's more important about peak engagement is the biological effect provoked, then that peak may be hours after the Cmax and--unless there is a specific reporter readout that's already been validated for the effect we care about--get completely missed in the effects of repeat dosing schedules. This is to say that the drug/perturbation has one attack curve, the response to it has another attack curve, and the relationship between those curves really really matters if we want perturbations to actually steer biological systems somewhere useful. Yes, we can carpet bomb patients with ongoing doses of a potent inhibitor and find that rheumatoid arthritis symptoms decrease when no cell in the body can respond to an IL6 signal, but this is not the same as understanding that there's a 5 day lag in tocilizumab suppressing that IL6 signal enough to rescue a patient from cytokine storm4. Notably, this was found out in an emergency setting, not proactively planned for. Despite the width and importance of gaps between perturbation and response attack curves, we tend to focus a lot more on characterizing the perturbation's attack curve than on its response and I suspect that there's a lot of really meaningful, directly translatable biology living in that gap, such as target recovery dynamics between daily doses or, for acutely stressful treatments like chemotherapy, background stress response adaptation to better survive the next dose5.
Going out of order to better introduce decay later, sustain ≈ steady state pharmacodynamics: if we keep a perturbation going, what response can the system maintain? Traditionally, we'd measure target inhibition or knockdown, which is still useful. However, this isn't the same as the actual sustained response because that sustained response is subject to its own resource constraints. There's an initial attack for that effect where extinguishing, say, STAT6, may cause an initial acute shock and fast reorganization in cells that survive it, but continuing that treatment can max out necessary cofactors or even recover activity levels. In cancer research we measure this as time to progression, where evolution beats treatment effects, but in other cases we often check in only rarely when I'm not so sure that we can trust that a perturbation's sustain levels are the same across every repeat exposure. Underlying conditions remodel, cells adapt, exogenous stressors unbalance things, and this shows up as continued disease progression despite ongoing treatment in chronic inflammation, cardiovascular disease, and neurodegeneration.
Release ≈ perturbation washout. What happens once the perturbation stops? Do its effects keep echoing through the cells for a long time or does it stop almost immediately? This has massive implications for a perturbation's actual effects on the system and we usually quit after getting a decent half life, leaving system recovery dynamics almost completely uncharacterized6. If (A) the perturbed system snaps right back after the drug washes out, it's memoryless and elastic. If (B) that system only drifts back to pre-treatment baselines slowly, there's an interesting viscous memory component worth characterizing7. And if (C) the perturbed system never returns to its pre-treatment baseline, then it has been fundamentally remodeled and should probably be treated as a new system. While we shoot for C with cell and gene therapies, studying the difference between A and B is largely reactive, only started after some population of patients have shown that drug discontinuation is not straightforwardly safe. There is almost no recovery data on treatments that have failed patients either. This mirrors the gap in data on insult recovery or salutogenesis. By focusing on what happens right after we give a drug and only counting the major categorical outcomes like disease progression, treatment discontinuation due to side effect profiles, or survival, we're missing a ton of release data and I wouldn't be surprised if there are useful treatable insights lurking in there waiting to be exploited. For example, if there's a 3 day surge in IL6 signaling after final methotrexate washout, there may be a subsequent 3 week exacerbation of rheumatoid arthritis symptoms due to all the indiscriminate inflammatory immune cell activation during washout, identifying a 3 day window in which an alternative inflammation-modulator (like ibuprofen or celecoxib) would provide an outsized patient benefit.
Decay ≈ the drop from attack peak to sustain levels while the perturbation is ongoing. This looks like a great initial response to a treatment followed by a rapid drop in its efficacy and it actually happens a lot more than we tend to measure routinely. For perturbation envelopes with fast attacks, that peak can be over in minutes as high concentrations saturate the target, but then its proximal effects have to ripple out to the rest of the affected biology where redundancy in function, cofactor depletion, or target downregulation can all lead to sharp decay curves. The term for this is tachyphylaxis and over a billion people experience it every single day with nicotine: the first ciggie of the morning feels amazing but by 1900 it's just a pacifier for withdrawal. Like smokers sleeping through the night, one solution is a perturbation holiday to reset the envelope, but that doesn't fix the underlying decay dynamics at play. It isn't limited to smoking, either, it also plays out in chemotherapy responses in cancer, opioid efficacy, anti-depressant efficacy, ADHD treatments, and more. I suspect it's also happening with ibuprofen but I have no direct evidence for it. Like release, I'm not sure that we can be sure decay curves are reliable between dosing schedules, but how much they vary across and across what territory again barely characterized.
We're still making billion dollar drug development investment decisions on metrics more crude and sparse than the modulation on a throwaway dubstep bassline to advertise dimensional lumber. Yes, a big chunk of this is due to expense and feasibility while the framework above is absolutely not shovel-ready. Not only are we missing data, we're also missing tools and I look forward to driving biostatisticians mad trying to model 7-component interaction terms on a decay curve surface as soon as possible. However, high-throughput characterization costs are also rapidly falling while drug development costs reach new stratospheric heights, so I am putting this out there as a sketch for where to prioritize looking for new data and how to treat it. By characterizing ADSR perturbation envelopes in new transgenic models we can understand if a model-critical edit has shifted cell physiology in a new and important way. By taking it into preclinical studies, we can start setting up proactive longitudinal response curve characterization and begin quantifying the coupling between 2 treatment's attack curves or the baseline shift of decay curves across studies to improve data comparability across development campaigns. And by modeling pathogenic trajectories via ADSR perturbation envelopes--potentially even in nested multi-omic formats instantiated individually per cell type to calculate cross-envelope coupling terms--we can begin to work more empirically with perturbation response surfaces and manifolds. These, in turn, let us understand how the treated systems evolve over time, whether a treatment will remain durable or be subject to tachyphylaxis limitations, and even identify more incisive molecular signals to measure in clinical trials. Although this is just a rough first draft for thinking through, the ADSR envelopes are everywhere in biology and we know it. We know that there isn't a single useful model of anything in biology that sits still when you perturb it. We know it's all dynamic, all the time, with interacting perturbations working across each one of the ADSR curves above. So while there's work to do in making these quantifiable and reliable, for now they're useful coherent parameters for thinking through perturbation effects and planning high-impact studies.
Yes yes yes, IC50 is not the right measurement for many biologics, cell, or gene therapies.
No promises they're nicely 2D or Euclidean though.
Perturbations: As I write this, the perturbations on my mind include pathophysiological processes, genetic manipulations, exogenous exposures, environmental insults, and model derivation. Probably not complete and likely to be some perturbation class-specific consequences that will need to be thought through.
Actually a cool bit of immunology, even if grim: cytokine storm that breaks out in severe COVID or some kinds of leukemia is treated with 1-2 doses of tocilizumab, a potent IL6R inhibitor that acts like a giant steam release valve for runaway inflammation. That treatment tamps down systemic immune freakout by telling all the circulating neutrophils in the body to go take a nap in the bone marrow and they do: calm down, relocate, tell other immune cells to take a deep breath with them.
I'd love to tackle methods for quantifying the coupling between perturbation and response in a large dataset of genetic KOs and multiplexed intrinsic stress response fluorescent reporters to get a handle on curve and coupling diversity, but I'm pretty sure that that dataset does not yet exist. Please hit me up if I'm wrong about that.
To be fair there's basically no funding for this, not grants, VC cash, or philanthropy.